
Ingestive Classics
Steve Fluharty and Alan Epstein on the control of salt appetite by angiotensin and aldosterone
FLUHARTY, STEVEN J, AND EPSTEIN, ALAN N.
Sodium appetite elicited by intracerebroventricular infusion of angiotensin II in the rat: II.
Synergistic interaction with systemic mineralocorticoids.
Behav Neurosci 97: 746-758, 1983 Oct;97(5):746-758.
Comment by Derek Daniels, October, 2018
When asked to contribute to the Ingestive Classics series, one paper immediately came to mind. My decision was obviously influenced by my training with Steve Fluharty, the fondness I have for my time in his lab, and the tremendous impact those years had on my thinking. But, every time I tried to think of a different paper, out of concern that my potential bias was a problem in my choice of a paper, the strength of the paper, the questions it raised, and the impact it has had on the field, strengthened my choice: the 1983 paper by Fluharty and Epstein [1], which provided the first direct experimental evidence for what came to be known as the “synergy hypothesis” of salt appetite.
Sodium appetite is a well-studied phenomenon in the field of ingestive behavior. It is an excellent example of a highly motivated behavior, and has served as a model system for the study of taste and taste-guided behaviors (see https://www.ssib.org/web/classic12.php for a previous Ingestive Classic about sodium taste). Fluharty and Epstein [1] approach the topic from a neuroendocrine perspective, and describe a set of experiments testing the hypothesis that angiotensin II (AngII) and aldosterone cooperate to drive intake of saline at a concentration not otherwise consumed by laboratory rats. The hypothesis was rooted in the firm understanding that the renin-angiotensin system, and the resultant adrenal release of aldosterone, were key in the physiology of sodium conservation. This role in sodium conservation was appreciated for years before scientists started to study the behavior that arises when rats are in a state of sodium need. Early attempts to extend what was known about sodium-conservation physiology to behavior found that administration of either AngII or aldosterone (or synthetic agonists) increased sodium appetite [2-7]. This was consistent with the hypothesis that the same hormones involved in sodium conservation were responsible for increased sodium appetite. Critical consideration of these findings, however, revealed that inducing a sodium appetite required a supraphysiological dose of either AngII or aldosterone [8-10]. Thus, it remained unclear how the renin-angiotensin-aldosterone system, so clearly important in the physiology of sodium conservation, affected behavior. This problem was solved, in part, by the demonstration of Fluharty and Epstein [1].
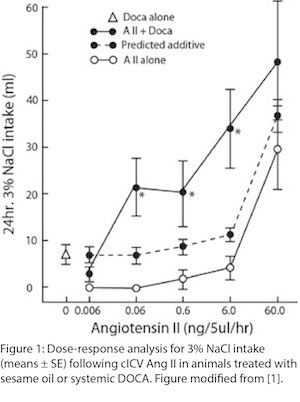
The experiments were relatively straightforward, with the key designs including a dose-response test of AngII on sodium intake with or without co-infusion of the synthetic mineralocorticoid, deoxycorticosterone acetate (DOCA), and a second experiment that gave small daily doses of DOCA to rats for a week before a similar dose-response test of AngII. In both experiments, the presence of DOCA (either given with the AngII or given chronically before AngII) led to greater sodium intake than did AngII alone, and did so with a magnitude greater than the sum of intake generated by AngII or DOCA alone (Figure 1), providing experimental evidence for the synergy of the peptide and the steroid. (It is noteworthy that the definition of “synergy” has been a topic of debate, here and elsewhere. In this instance, it was defined as an effect generated by the combination of treatments that is greater than would be predicted by summing the effects of each treatment when given alone. Although this is no longer considered a valid synergy criterion, the data in Figure 1 suggest that some current criteria were satisfied [11-13].
The demonstration of synergy was remarkable, and remains important today for several reasons, including that it provided a framework for future study. Subsequent studies showed a converse effect: that blocking either AngII or aldosterone produced only a partial attenuation of sodium intake by sodium deprived rats, but blocking both AngII and aldosterone caused a complete prevention of intake [14]. Other studies found that the synergy effect also occurred in other species, including baboons [15]. Finally, thirty-four years after the report by Fluharty and Epstein, Resch et al. [16] used chemogenetic approaches to identify the ventral bed nucleus of the stria terminalis as a site of interaction between aldosterone-sensitive neurons with cell bodies in the nucleus of the solitary tract and AngII-sensitive neurons with dell bodies in the subfornical organ. This exciting advance in our understanding of the neural control of sodium appetite arose directly from Fluharty and Epstein’s classic study.
The excellent work that arose from the paper may be enough to make it an “ingestive classic,” but the major significance of this paper, in my view, lies also in the resolution it provided to a vexing problem in the study of sodium appetite at the time it was published. Key in this solution is the importance that Fluharty and Epstein emphasized in understanding and considering the whole animal when performing experimental manipulations of single substrates. Indeed, before these experiments, as briefly described above, the idea that AngII played a key role in sodium appetite faced several challenges. These challenges included arguments that physiological doses of AngII did not generate sodium appetite [9, 10], and that it was not clear if the appetite was a direct function of AngII, or more directly related to AngII-induced natriuresis [17, 18]. It is now clear that part of the problem stemmed from attempting to draw conclusions about these hormones based on experiments using “normal” rats; rats that would not normally experience elevated levels of these hormones without other relevant changes in their physiology. In other words, the report highlighted that an otherwise untreated laboratory rat is at a very different starting point than a rat would be when endogenous AngII and aldosterone are elevated. This is because endogenous AngII and aldosterone are elevated in response to challenges to fluid homeostasis, such as sodium depletion and hypovolemia. That creates a very different baseline in many ways. For instance, in an otherwise untreated rat, administration of AngII generates an acute pressor response. But a hypovolemic rat, in which the endogenous renin-angiotensin-aldosterone system would be engaged, is also hypotensive, so the pressor response raises blood pressure to normotensive levels, rather than causing acute hypertension. This is especially important with respect to intake, because elevated blood pressure inhibits intake via arterial baroreceptors [19, 20]. Thus, when AngII is given to an otherwise normal rat, the intake-stimulating effects of AngII (with or without aldosterone) are partially tempered by the concomitant intake-suppressing effect of the acute hypertension caused by the treatment. Likewise, with respect to the relationship between AngII and aldosterone, the studies of Fluharty and Epstein highlighted the fact that rats more naturally driven to consume sodium would never experience elevated AngII in isolation, but would experience it accompanied by elevated aldosterone. Although this is described and discussed by Fluharty and Epstein as related to their work on AngII and aldosterone, the lesson of considering the physiological context of the whole animal when testing the effect of any isolated stimuli remains important. This is an important lesson for many of us.
References
[1] Fluharty, S. J., Epstein, A. N. Sodium appetite elicited by intracerebroventricular infusion of angiotensin II in the rat: II. Synergistic interaction with systemic mineralocorticoids. Behavioral Neuroscience. 1983,97:746-58.
[2] Wolf, G., Handal, P. J. Aldosterone-induced sodium appetite: dose-response and specificity. Endocrinology. 1966,78:1120-4.
[3] Findlay, A. L., Epstein, A. N. Increased sodium intake is somehow induced in rats by intravenous angiotensin II. Hormones and Behavior. 1980,14:86-92.
[4] Avrith, D. B., Fitzsimons, J. T. Increased sodium appetite in the rat induced by intracranial administration of components of the renin-angiotensin system. The Journal of Physiology. 1980,301:349-64.
[5] Avrith, D. B., Wiselka, M. J., Fitzsimons, J. T. Increased sodium appetite in adrenalectomized or hypophysectomized rats after intracranial injections of renin or angiotensin II. The Journal of Endocrinology. 1980,87:109-12.
[6] Bryant, R. W., Epstein, A. N., Fitzsimons, J. T., Fluharty, S. J. Arousal of a specific and persistent sodium appetite in the rat with continuous intracerebroventricular infusion of angiotensin II. The Journal of Physiology. 1980,301:365-82.
[7] Rice, K. K., Richter, C. P. Increased sodium chloride and water intake of normal rats treated with desoxycorticosterone acetate. Endocrinology. 1943,33:106-15.
[8] Fregley, M. J., Waters, I. W. Effect of mineralocorticoids on spontaneous sodium chloride appetite of adrenalectomized rats. Physiology & Behavior. 1966,1:65-74.
[9] Ramsay, D. J. The brain renin angiotensin system: a re-evaluation. Neuroscience. 1979,4:313-21.
[10] Stricker, E. M., Vagnucci, A. H., McDonald, R. H., Jr., Leenen, F. H. Renin and aldosterone secretions during hypovolemia in rats: relation to NaCl intake. The American Journal of Physiology. 1979,237:R45-51.
[11] Geary, N. Understanding synergy. American Journal of Physiology - Endocrinology and Metabolism. 2013,304:E237-53.
[12] Lederer, S., Dijkstra, T. M. H., Heskes, T. Additive Dose Response Models: Explicit Formulation and the Loewe Additivity Consistency Condition. Frontiers in Pharmacology. 2018,9:31.
[13] Foucquier, J., Guedj, M. Analysis of drug combinations: current methodological landscape. Pharmacology Research & Perspectives. 2015,3:e00149.
[14] Sakai, R. R., Nicolaidis, S., Epstein, A. N. Salt appetite is suppressed by interference with angiotensin II and aldosterone. The American Journal of Physiology. 1986,251:R762-8.
[15] Shade, R. E., Blair-West, J. R., Carey, K. D., Madden, L. J., Weisinger, R. S., Denton, D. A. Synergy between angiotensin and aldosterone in evoking sodium appetite in baboons. American Journal of Physiology - Regulatory, Integrative and Comparative Physiology. 2002,283:R1070-8.
[16] Resch, J. M., Fenselau, H., Madara, J. C., Wu, C., Campbell, J. N., Lyubetskaya, A., et al. Aldosterone-Sensing Neurons in the NTS Exhibit State-Dependent Pacemaker Activity and Drive Sodium Appetite via Synergy with Angiotensin II Signaling. Neuron. 2017,96:190-206 e7.
[17] Avrith, D. B., Fitzsimons, J. T., Nicolaidis, S. The effects of longterm intravenous infusions of angiotensin II on thirst, sodium appetite, and water and sodium balance in the rat. Proceedings of the VII International Commission of the Physiology of Food and Fluid Intake. 1980.
[18] Fluharty, S. J., Manaker, S. Sodium appetite elicited by intracerebroventricular infusion of angiotensin II in the rat: I. Relation to urinary sodium excretion. Behavioral Neuroscience. 1983,97:738-45.
[19] Thunhorst, R. L., Johnson, A. K. Effects of arterial pressure on drinking and urinary responses to intracerebroventricular angiotensin II. The American Journal of Physiology. 1993,264:R211-7.,
[20] Thunhorst, R. L., Lewis, S. J., Johnson, A. K. Role of arterial baroreceptor input on thirst and urinary responses to intracerebroventricular angiotensin II. The American Journal of Physiology. 1993,265:R591-5.